Dynamical fingerprints: A theoretical framework for understanding biomolecular processes by combination of simulation and kinetic experiments
/
Frank Noé, Sören Doose, Isabella Daidone, Marc Löllmann, Markus Sauer, John D. Chodera, and Jeremy C. Smith.
Proc. Natl. Acad. Sci. USA 108:4822, 2011. [DOI] [PDF]
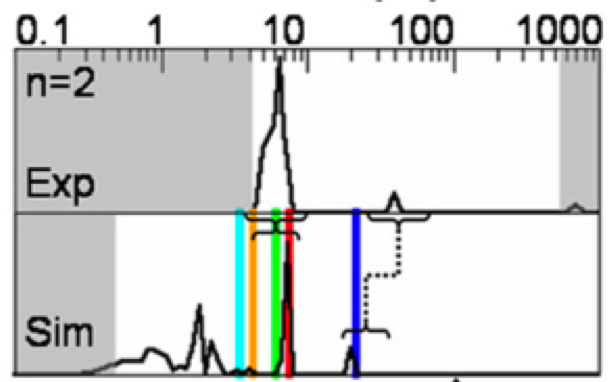
We present a new framework for comparing essential features of the dynamics between experiment and simulation to identify the kinetics processes contributing to individual relaxation timescales in perturbation-response or correlation spectroscopy experiments.