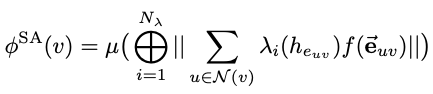NNP/MM: Fast molecular dynamics simulations with machine learning potentials and molecular mechanics
/
Galvelis R, Varela-Rial A, Doerr S, Fino R, Eastman P, Markland TE, Chodera JD, and de Fabritiis G
Journal of Chemical Information and Modeling 63:5701, 2023 [DOI] [arXiv]
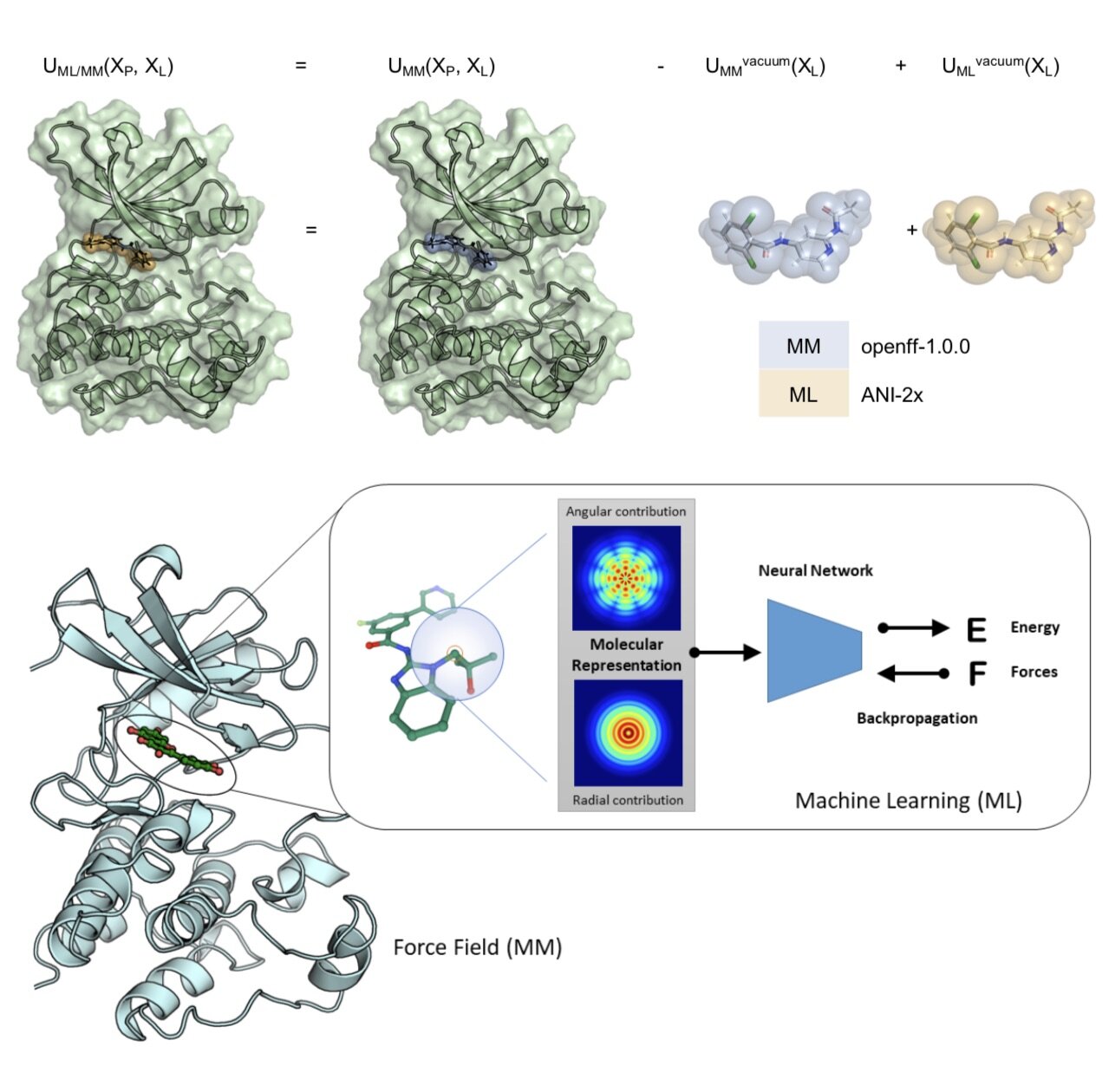
We demonstrate that a new generation of quantum machine learning (QML) potentials based on neural networks---which can achieve quantum chemical accuracy at a fraction of the cost---can be implemented efficiently in the OpenMM molecular dynamics simulation engine as part of hybrid machine learning / molecular mechanics (ML/MM) potentials that promise to deliver superior accuracy for modeling protein-ligand interactions.