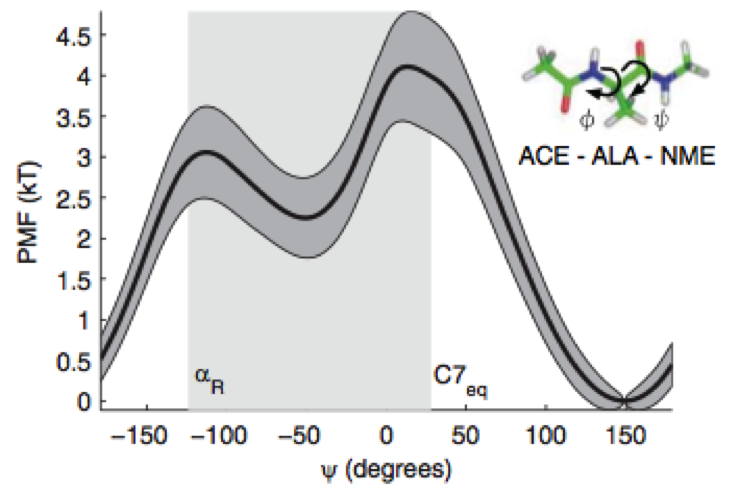
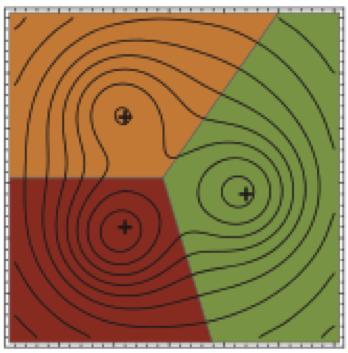
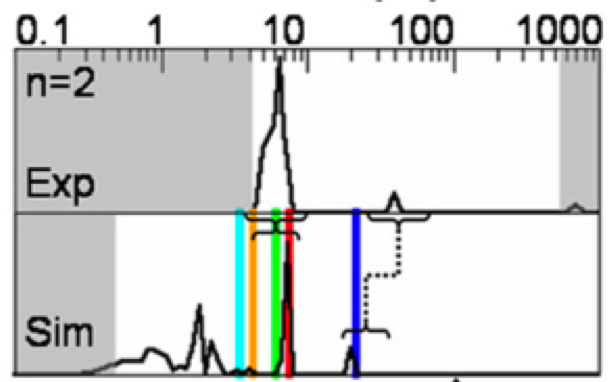
Spectral rate theory for two-state kinetics
/
Jan-Hendrik Prinz, John D. Chodera, and Frank Noé.
Phys. Rev. X 4:011020, 2014. [DOI] [PDF]
We present a new mathematical framework for unifying various two-state rate theories presented in the physical chemistry literature over many decades, and provide a quantitative way to measure reaction coordinate quality.