
RESEARCH
Featured
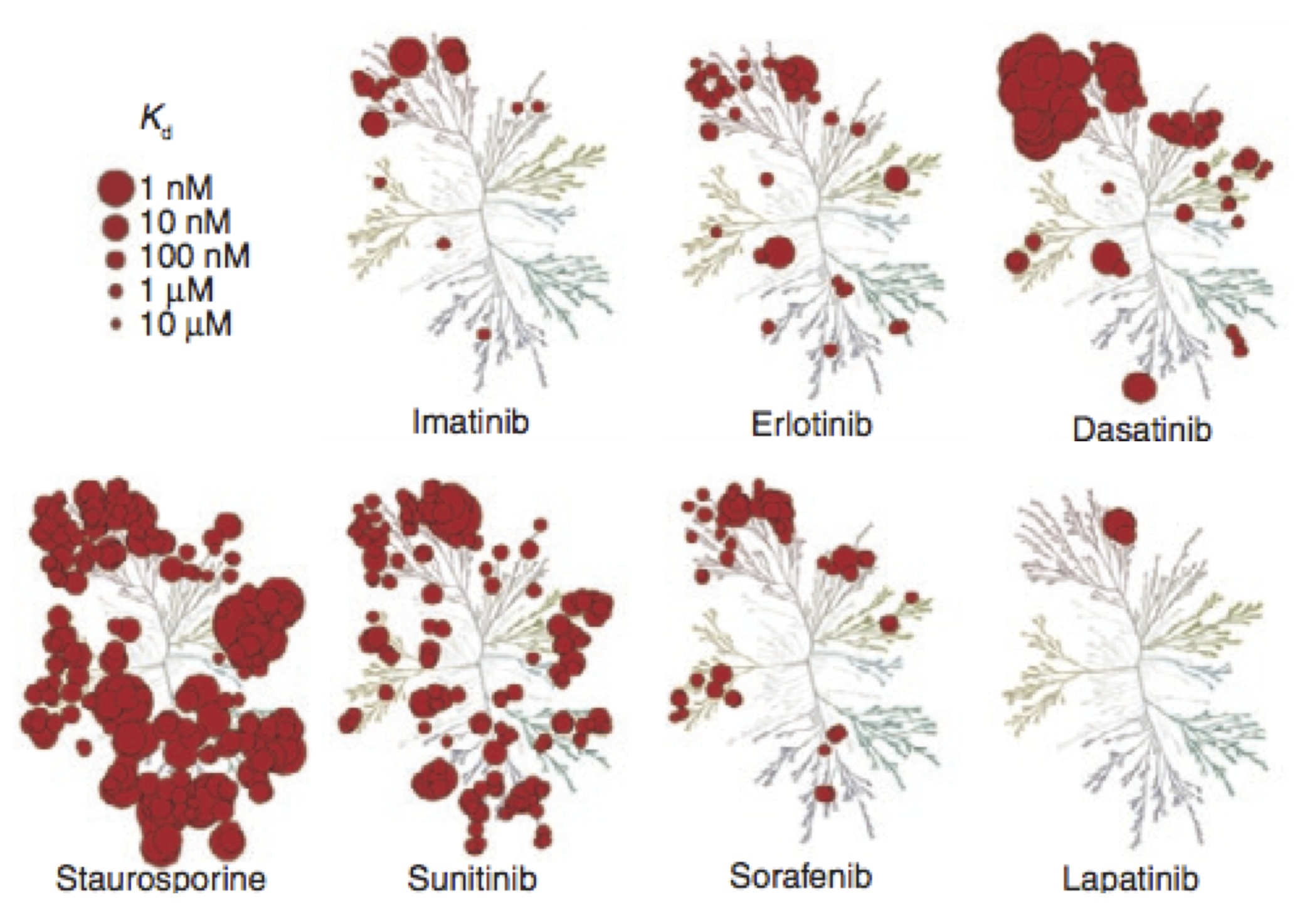






Selective kinase inhibitors---such as the blockbuster drug imatinib---have shown tremendous promise in the treatment of cancers involving kinase dysregulation. Currently, over 27 small molecule targeted kinase inhibitors have received FDA approval, representing a substantial fraction of the $37B U.S.~market for oncology drugs. Despite this, major challenges remain in their widespread application in cancer treatment. To meet these challenges, our laboratory develops quantitative physical models of kinase inhibitor efficacy to accelerate the rational design of kinase inhibitors with desired selectivity profiles, an understanding of mutational mechanisms of resistance, and prediction of drug sensitivity and resistance in individual patient tumors.
While there are now over 30 FDA-approved selective kinase inhibitors available for the treatment of cancer, the median progression-free survival is still <1 year for a majority of these drugs. Drug resistance is responsible for >90% of deaths in patients with metastatic cancer. In many of these cases, mutations in the target of therapy drive resistance by abolishing or reducing inhibitor affinity while maintaining or increasing kinase activity.
Funded by a STARR Cancer Consortium grant with Minkui Luo, we are working to identify hidden conformations of protein methyltransferases to aid the Luo lab in developing useful chemical probes for elcudiating the functions of these important epigenetic disease targets.
The Heller lab at MSKCC has discovered that poorly soluble kinase inhibitors mixed with specific indocyanine dye excipients will spontaneously form nanoparticles with very high (90% by mass) drug loadings, and that these dyes specifically target certain tumors while maintaining high blood stability. These nanoparticles offer the potential for avoiding both off- and on-pathway toxicities while delivering high quantities of targeted kinase inhibitors directly to tumors.
We have developed an enhanced sampling approach that allows us to explore combinatorially large spaces of inhibitor designs in a way that automatically biases the simulation toward inhibitors with higher affinity for one or more targets. This approach---based on expanded ensemble simulations and made possible by a new nonequilibrium Monte Carlo algorithm we developed---promises to provide a time- and cost-effective solution to the problem of optimizing small molecules for affinity. By restricting the space of compounds to those accessible from a given set of commercially-available starting materials and a library of common synthetic transformations, we aim to propose a set of compounds that have a high likelihood of increased potency and are likely to be readily synthesizable.
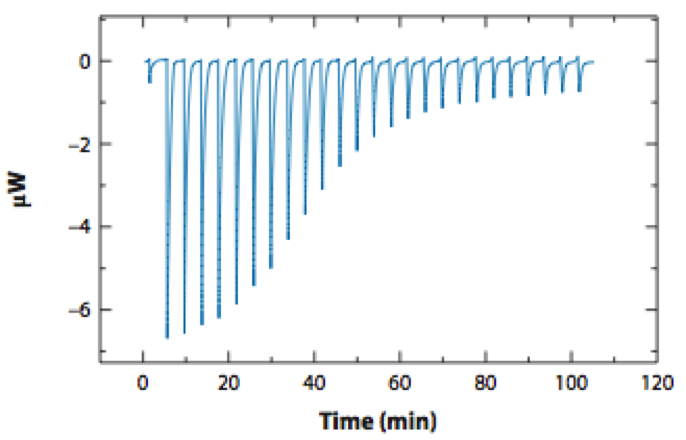
To drive improvements in quantitative accuracy, we use automated biophysical experiments to probe the physical determinants of small molecule affinity and selectivity. Using robotically driven site-directed mutagenesis to perturb the protein, rather than synthesize new small molecules, we can rapidly collect data to improve algorithms, forcefields, and the treatment of chemical effects in protein-ligand modeling, as well as address fundamental physical questions about what interactions are critical in determining small molecule affinity and selectivity.
Ras family proteins, important in the control of cell growth via signaling, are commonly mutated in human cancer. Activating mutations in Ras are a leading cause of resistance to modern targeted therapy, and patients who harbor Ras mutations have considerably poorer prognoses than those with wild type Ras. Targeting Ras has proven difficult because oncogenic mutations activate Ras primarily by ablating enzymatic activity, leaving classical enzyme inhibition strategies unworkable. The high affinity of Ras for GTP---which locks Ras in an active conformation---combined with high intracellular GTP concentrations makes outcompeting the bound nucleotide extremely difficult.
All experimental assay data contains error arising from uncertainties in initial compositions, dispensed masses or volumes, measurement noise, model fitting error, and intrinsic biological variability. Accounting for this error to produce a reliable estimate of the uncertainty of experimentally-derived quantities is critical, as this is the basis for testing hypotheses or building predictive models, but it is often difficult to even identify the dominant sources of assay error, let alone propagate them. Our lab uses two primary tools to both build predictive models of assay error and incorporate all sources of error and uncertainty in data analysis: the bootstrap principle and Bayesian inference.
To enable truly rational computational design of small molecules, we are developing new algorithms and open source tools for alchemical free energy calculations that provide a rigorous but practical approach to the quantitative prediction of small molecule binding affinities and other physicochemical properties of relevance to ADME-Tox (such as partition coefficients, serum binding, and off-target affinities).