Development and benchmarking of Open Force Field 2.0.0---the Sage small molecule force field
/
Boothroyd S, Behara PK, Madin OC, Hahn DF, Jang H, Gapsys V, Wagner JR, Horton JT, Dotson DL, Thompson MW, Maat J, Gokey T, Wang L-P, Cole DJ, Gilson MK, Chodera JD, Bayly CI, Shirts MR, Mobley DL
Journal of Chemical Theory and Computation 19:3251, 2023 [DOI] [chemRxiv] [GitHub] [examples]
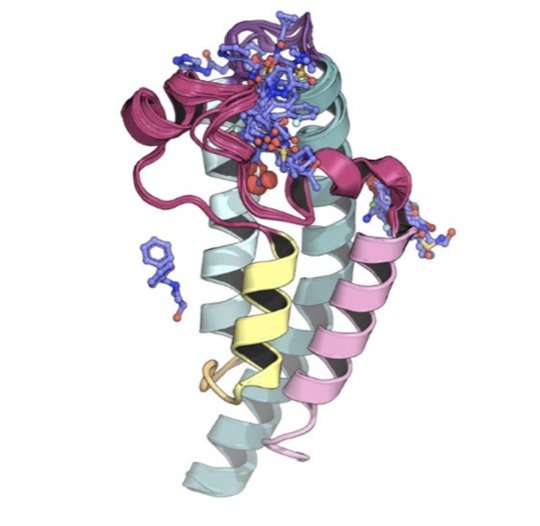
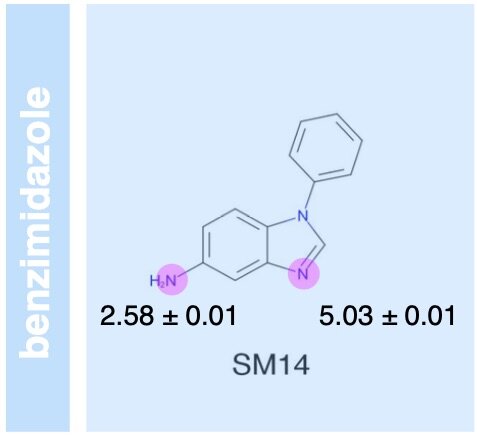
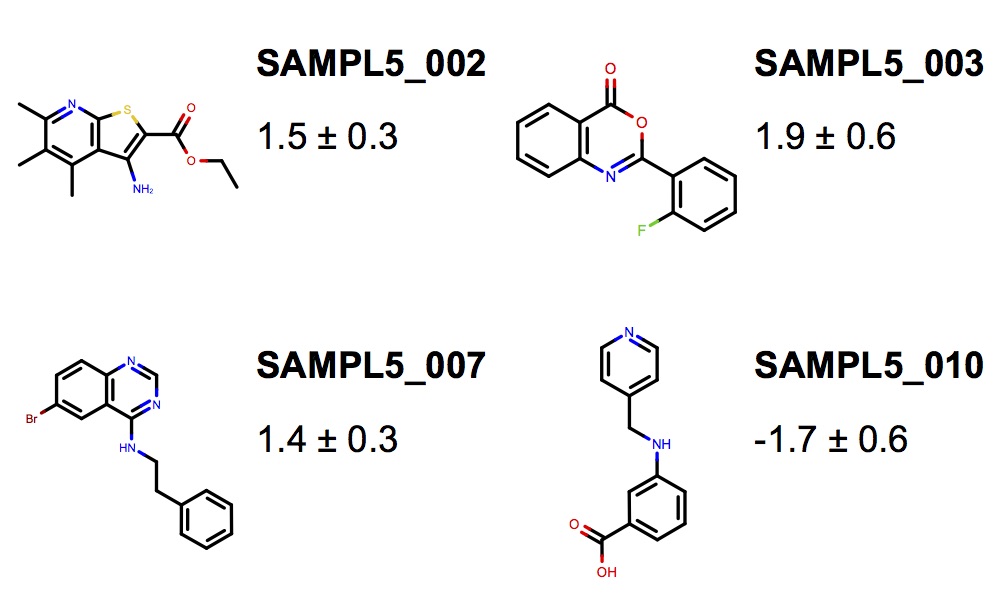
We present a new generation of small molecule force field for molecular design from the Open Force Field Initiative fit to both quantum chemical and experimental liquid mixture data