Fitting quantum machine learning potentials to experimental free energy data: Predicting tautomer ratios in solution
/
Marcus Wieder, Josh Fass, and John D. Chodera
Chemical Science, in press [bioRxiv] [code]
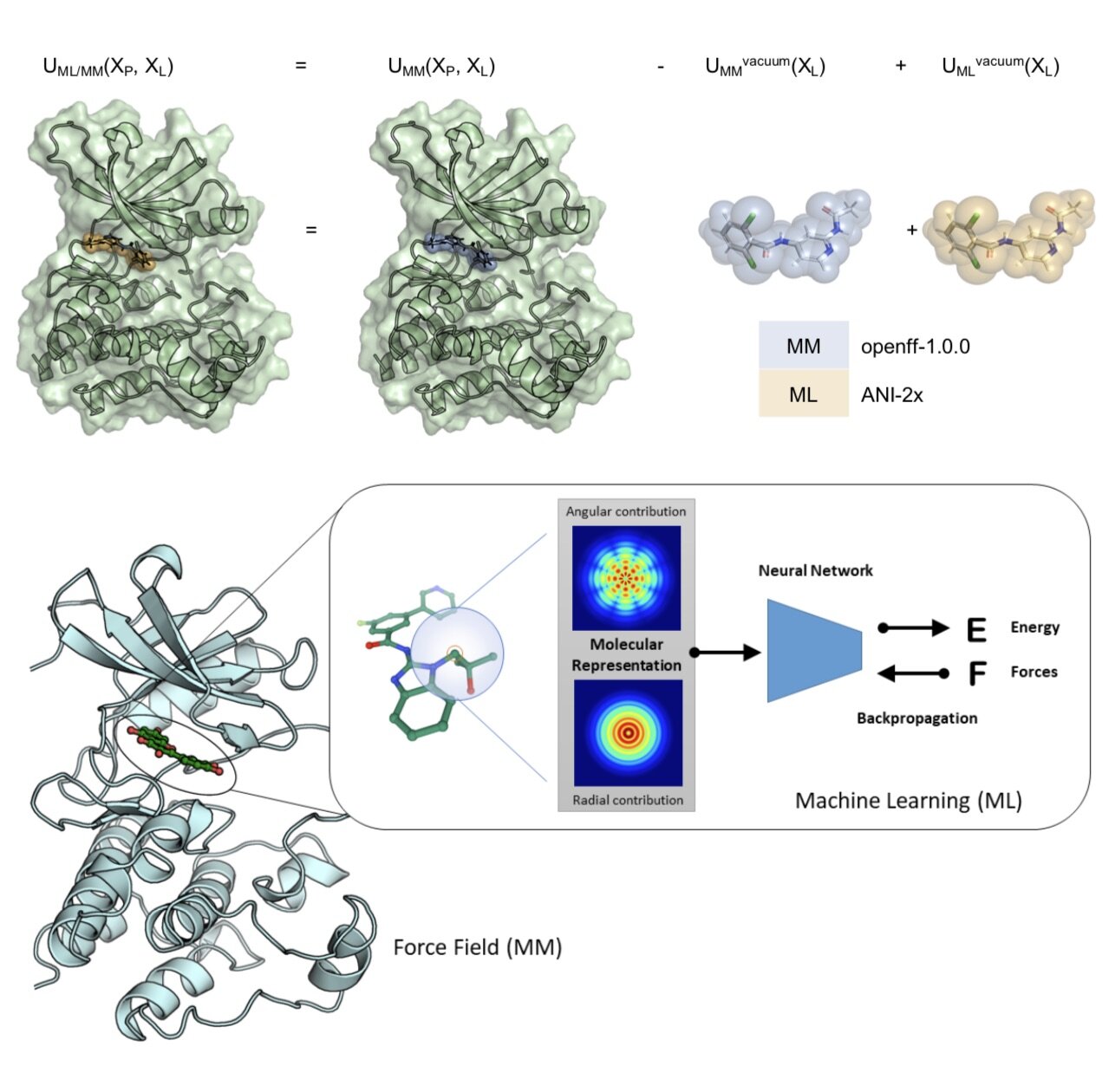
We demonstrate, for the first time, how alchemical free energy calculations can performed on systems simulated entirely with quantum machine learning potentials and how these potentials can be retrained on experimental free energies to generalize to new molecules from limited training data. We apply this approach to a difficult problem in small molecule drug discovery: Predicting accurate tautomer ratios in solution.