Kinase inhibitor selectivity and design
/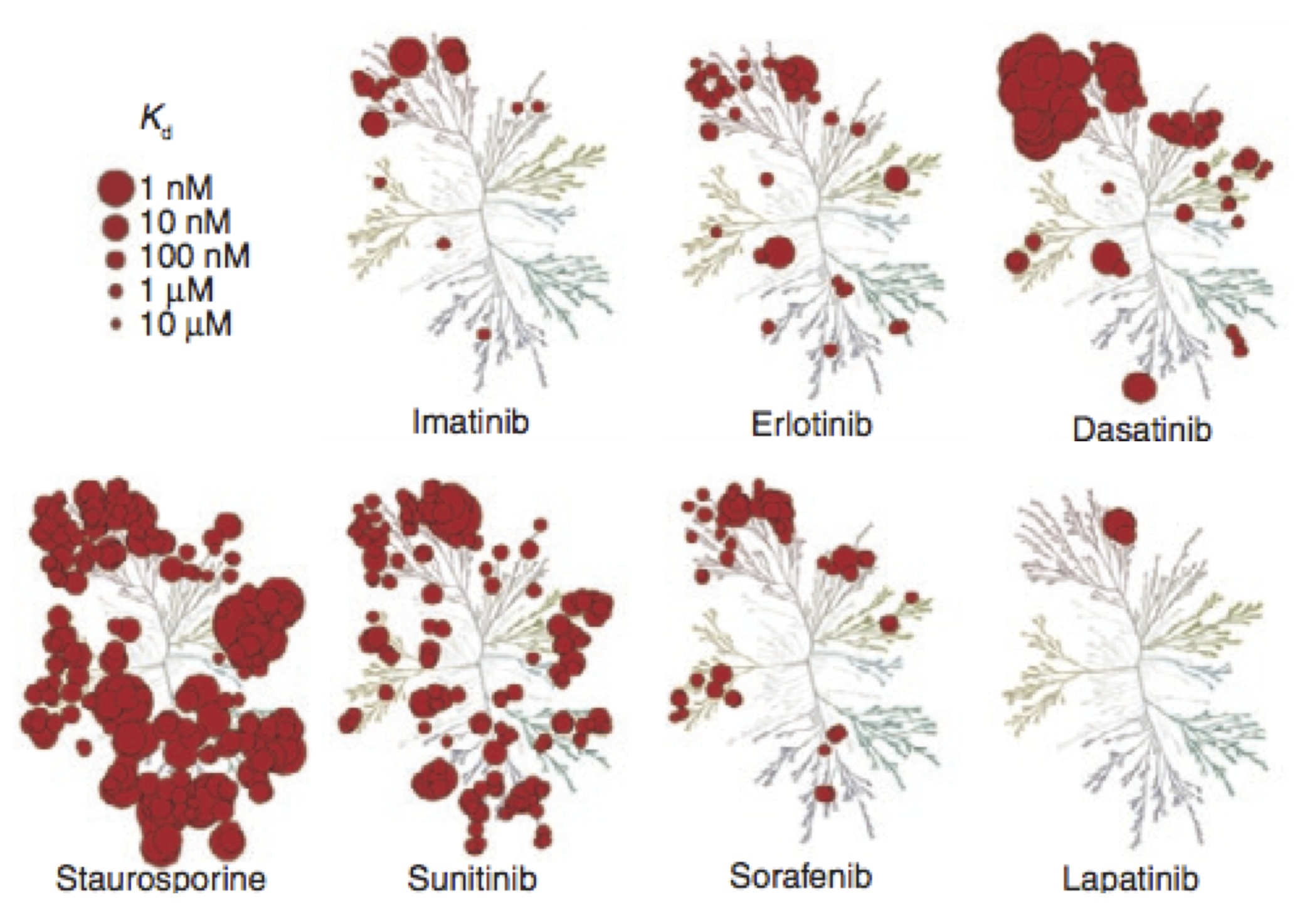
From Nature Biotech. 23:329, 2005.
Selective kinase inhibitors---such as the blockbuster drug imatinib---have shown tremendous promise in the treatment of cancers involving kinase dysregulation. Currently, over 27 small molecule targeted kinase inhibitors have received FDA approval, representing a substantial fraction of the $37B U.S.~market for oncology drugs. Despite this, major challenges remain in their widespread application in cancer treatment.

First, the design of potent inhibitors with desired selectivity profiles is difficult and expensive; existing approaches---based on screening targeted libraries followed by rounds of structure-guided lead optimization---generally require several years and hundreds of millions of dollars to produce a clinical candidate. Second, these molecules are still generally poorly selective, with the risk that off-target effects will limit therapeutic index. Third, the often-rapid emergence of drug resistance can eliminate therapeutic benefit; over 90% of metastatic cancer deaths are believed to result from drug-resistant forms. To meet these challenges, our laboratory develops quantitative physical models of kinase inhibitor efficacy to accelerate the rational design of kinase inhibitors with desired selectivity profiles, an understanding of mutational mechanisms of resistance, and prediction of drug sensitivity and resistance in individual patient tumors.
DESIGN OF KINASE INHIBITORS WITH DESIRED SELECTIVITIES
Recent evidence suggests that a major effect in the selectivity of imatinib for Abl over the closely-related Src is the larger conformational reorganization energetic cost that Src must pay in order to adopt a binding-competent conformation. While failure to account for this contribution will frustrate the ability rationally engineer kinase inhibitors with desired selectivity profiles, it is difficult to measure these reorganization energies experimentally.
As part of the Folding@home Consortium, we use massively parallel molecular dynamics simulations on the Folding@home worldwide distributed computing platform---the planet's largest computer for biology, with aggregate performance of over 100 PFLOP/s---to computationally map the conformations accessible to kinases and their associated energetics. We have developed a tool Ensembler to automate the modeling of human kinase catalytic domains onto all available kinase structures to broadly capture the variety of active and inactive conformations available to kinases, which are well-known to be highly conformationally heterogeneous. We then employ the Markov state modeling (MSM) approach we originally developed (in collaborations with the Pande lab, the Noé group, and IBM Almaden Research) to study protein folding to extract distinct conformational states, energetics, and activation kinetics. We are currently studying a number of kinases of interest to cancer and other diseases---ABL, SRC, MEK, MTOR, AURKA, DDR1, CK2, and SYK---and are in the process of extending this approach to all human kinases.
To exploit these conformational and energetic maps of kinase conformations to understand kinase inhibitor affinity and selectivity, as well as facilitate the engineering of new selective inhibitors, we have developed a new generation of quantitatively accurate physical models based on alchemical free energy calculations. These methods use molecular mechanics forcefields to rigorously compute binding affinities that include all relevant statistical mechanical effects, including entropic and enthalpic contributions. To make these techniques fast enough for practical use, we have built a new open-source GPU-accelerated code YANK.

In addition to allowing for accurate inclusion of the reorganization energy for different kinases, our kinase MSMs also reveal opportunities for allosterically targeting kinases by identifying conformations that expose otherwise cryptic binding sites that are not visible in crystallographic structures unless an allosteric inhibitor has already been identified and cocrystallized. As few allosteric kinase inhibitors have been identified to date, this represents an emerging opportunity for engineering new classes of effective therapeutics for cancer.
ACCELERATED SAMPLING OF KINASE CONFORMATIONAL STATES
While MSMs built on Folding@home are useful in identifying accessible kinase conformations and energetics, these models often require multiple milliseconds of aggregate simulation time, making them incredibly expensive to generate. We are experimenting with accelerated sampling schemes based on new machine learning techniques for for learning slow degrees of freedom from molecular simulations (such as VAMPnets, SGOOP, AMINO, and RAVE) to learn kinase superfamily specific order parameters and combine them with alchemical free energy calculations.
SOFTWARE
ENSEMBLER: An tool for automated modeling and simulation at the superfamily scale
RESOURCES
Explore a list of kinases that have been expressed in E. coli constructed by postdoc Daniel L. Parton. Plasmids are made available via AddGene.
A prioritized list of kinase catalytic domains for our own cloning and expression experiments in E. coli
COLLABORATORS
Markus Seeliger (Stony Brook University): Kinase biochemistry and structural biology
Nicholas Levinson (University of Minnesota): Kinase biochemistry, spectroscopy, and structural biology
Frank Noé (Freie Universität Berlin): Markov state models for biomolecular conformational dynamics
Vijay S. Pande (Stanford University): Markov state models for biomolecular conformational dynamics
PERSONNEL
Sonya M. Hanson (postdoctoral fellow): Reorganization energy and conformational dynamics of kinases; automated experimental assays
Steven A. Albanese (Gerstner Sloan Kettering Graduate Student): Structural and energetic ramifications of kinase mutants
SELECTED PUBLICATIONS
Ensembler: Enabling high-throughput molecular simulations at the superfamily scale
Daniel L. Parton, Patrick B. Grinaway, Sonya M. Hanson, Kyle A. Beauchamp, and John D. Chodera
PLoS Comput. Biol. 12:e1004728, 2016. [DOI] [PDF] [bioRxiv] / data: [Dryad] / code: [GitHub]
An open library of human kinase domain constructs for automated bacterial expression
Daniel L. Parton, Sonya M. Hanson, Lucelenie Rodríguez-Laureano, Steven K. Albanese, Scott Gradia, Chris Jeans, Markus Seeliger, and John D. Chodera.
Manuscript prior to publication: [bioRxiv] / interactive data browser: [github.io] / plasmids available via AddGene
Identifying ligand binding sites and poses using GPU-accelerated Hamiltonian replica exchange molecular dynamics
Kai Wang K, John D. Chodera, Yanzhi Yang, and Michael R. Shirts.
J. Comput. Aid. Mol. Des. 27:989, 2013. [DOI] [PDF]
Confine-and-release method: Obtaining correct binding free energies in the presence of protein conformational change
David L. Mobley, John D. Chodera, and Ken A. Dill.
J. Chem. Theor. Comput. 3:1231, 2007. [DOI] [PDF]
Automatic discovery of metastable states for the construction of Markov models of macromolecular conformational dynamics
John D. Chodera*, Nina Singhal*, William C. Swope, Jed W. Pitera, Vijay S. Pande, and Ken A. Dill.
J. Chem. Phys. 126:155101, 2007. [DOI] [PDF]
FUNDING
NIGMS 1 R01 GM121505 01